2026
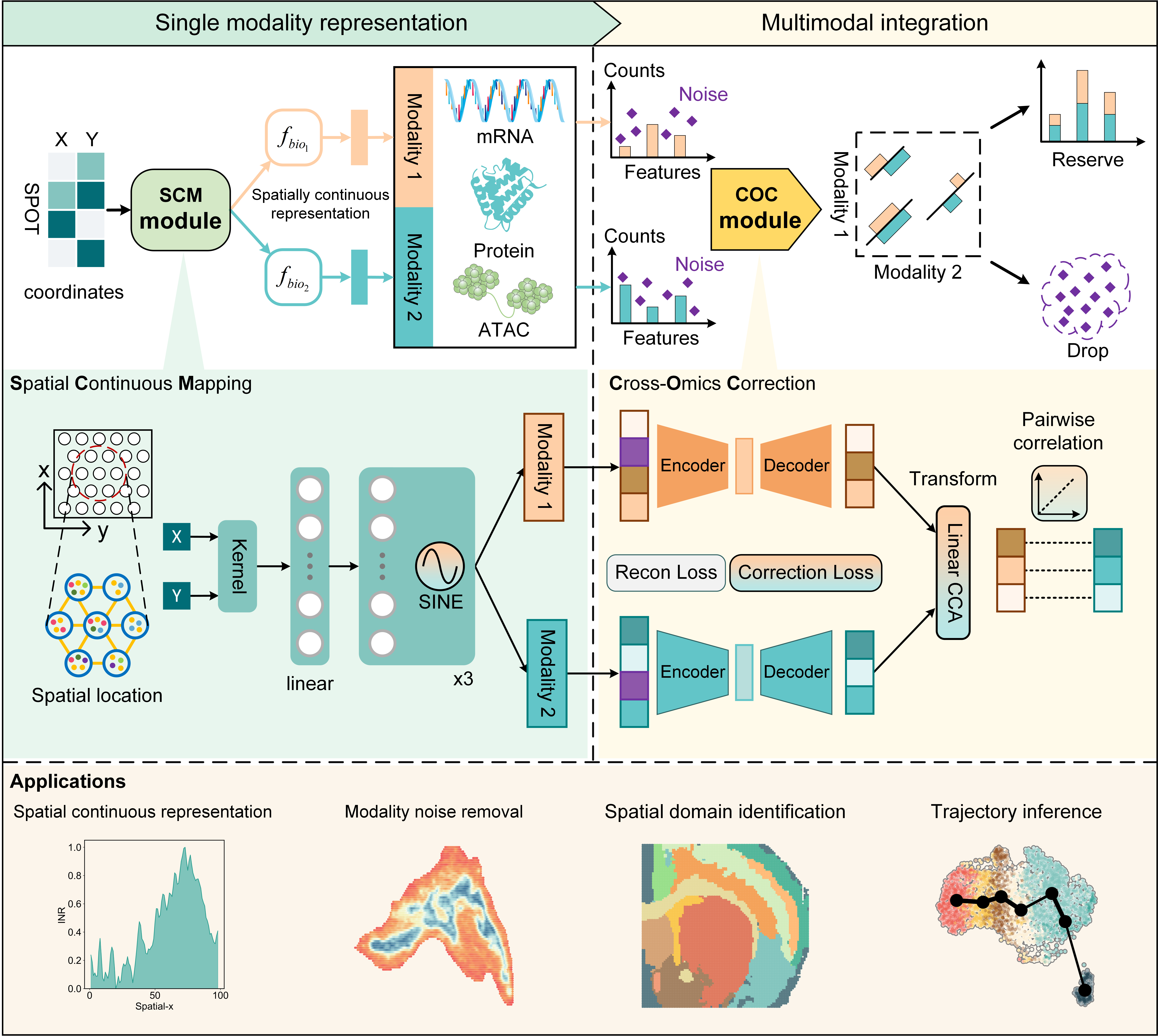
SpatialCOC: an integrative framework for spatial continuous mapping and cross-omics correction in spatial multi-omics data
Mingxuan Li (黎明轩)*, Peisen Sun (孙培森)*, Yisi Luo (罗倚斯), Guancheng Zhou (周冠程), Xiaofei Yang (杨晓飞), Deyu Meng (孟德宇), Kai Ye (叶凯) (* equal contribution)
Nature Communications 2026
Integrating spatial multi-omics data presents significant challenges, particularly in uncovering the spatial patterns of cells and deciphering the real regulatory mechanisms among various omics. These insights are critical for harnessing the full potential of each modality while minimizing the impact of biotechnological biases that will lead to unstable results. Here, we introduce SpatialCOC, a framework that treats spatial information as prior knowledge to learn omics-specific spatial distributions, then discovering nonlinear correlations among modalities. The effectiveness and robustness of SpatialCOC are validated using real-world datasets, encompassing diverse tissue sections analyzed with multiple experimental techniques. Compared to existing methods, SpatialCOC excels in identifying region-specific continuous spatial domains and maintains batch-consistency across trajectory inferences. By providing a novel perspective on the interplay between spatial information and multi-omics modalities, SpatialCOC offers a flexible approach that can accommodate modality data of arbitrary dimensions.
SpatialCOC: an integrative framework for spatial continuous mapping and cross-omics correction in spatial multi-omics data
Mingxuan Li (黎明轩)*, Peisen Sun (孙培森)*, Yisi Luo (罗倚斯), Guancheng Zhou (周冠程), Xiaofei Yang (杨晓飞), Deyu Meng (孟德宇), Kai Ye (叶凯) (* equal contribution)
Nature Communications 2026
Integrating spatial multi-omics data presents significant challenges, particularly in uncovering the spatial patterns of cells and deciphering the real regulatory mechanisms among various omics. These insights are critical for harnessing the full potential of each modality while minimizing the impact of biotechnological biases that will lead to unstable results. Here, we introduce SpatialCOC, a framework that treats spatial information as prior knowledge to learn omics-specific spatial distributions, then discovering nonlinear correlations among modalities. The effectiveness and robustness of SpatialCOC are validated using real-world datasets, encompassing diverse tissue sections analyzed with multiple experimental techniques. Compared to existing methods, SpatialCOC excels in identifying region-specific continuous spatial domains and maintains batch-consistency across trajectory inferences. By providing a novel perspective on the interplay between spatial information and multi-omics modalities, SpatialCOC offers a flexible approach that can accommodate modality data of arbitrary dimensions.
2025
Spatially resolved single-cell transcriptome analysis of murine Salmonella infection reveals the role of distal colonocytes in the inflammatory response
Dan Xu, Ruifen Zhang, Shanshan Li, Can Guo, Chenglin Guan, Xiang Li, Mengyao Guo, Xin Xu, Yaxin Liu, Chenyi Mao, Peisen Sun, Xiaomin Dang, Diya Sun, Chengyao Wang, Stephen J. Bush, Kai Ye
Gut Microbes 2025
The intestine is a highly compartmentalized organ, with distinct segments exhibiting both varying susceptibilities and responses to enteric pathogens, although the cellular and molecular bases of these responses remain elusive. Here, we used Salmonella Typhimurium (S. Tm), a prominent enteric pathogen that causes human colitis, to establish a murine model of Salmonella enterocolitis. By integrating bulk RNA-seq, single-cell RNA-seq, and spatial RNA-seq data, we present a comprehensive spatiotemporal single-cell transcriptomic landscape of the colon over a week-long time course of infection. We identified the distal colon as the intestinal segment where most of the host responses were initiated, with distal colonocytes (DCCs) being the most responsive epithelial cells upon the onset of infection. Furthermore, by correlating our findings with human intestinal single-cell transcriptome data, we identified a human colonocyte population that shares many characteristics with murine DCCs. Our study advances the understanding of the cellular and molecular basis of compartmentalized intestinal responses to pathogenic insults and may pave the way for novel preventive and therapeutic strategies to mitigate intestinal damage and combat intestinal infections.
Spatially resolved single-cell transcriptome analysis of murine Salmonella infection reveals the role of distal colonocytes in the inflammatory response
Dan Xu, Ruifen Zhang, Shanshan Li, Can Guo, Chenglin Guan, Xiang Li, Mengyao Guo, Xin Xu, Yaxin Liu, Chenyi Mao, Peisen Sun, Xiaomin Dang, Diya Sun, Chengyao Wang, Stephen J. Bush, Kai Ye
Gut Microbes 2025
The intestine is a highly compartmentalized organ, with distinct segments exhibiting both varying susceptibilities and responses to enteric pathogens, although the cellular and molecular bases of these responses remain elusive. Here, we used Salmonella Typhimurium (S. Tm), a prominent enteric pathogen that causes human colitis, to establish a murine model of Salmonella enterocolitis. By integrating bulk RNA-seq, single-cell RNA-seq, and spatial RNA-seq data, we present a comprehensive spatiotemporal single-cell transcriptomic landscape of the colon over a week-long time course of infection. We identified the distal colon as the intestinal segment where most of the host responses were initiated, with distal colonocytes (DCCs) being the most responsive epithelial cells upon the onset of infection. Furthermore, by correlating our findings with human intestinal single-cell transcriptome data, we identified a human colonocyte population that shares many characteristics with murine DCCs. Our study advances the understanding of the cellular and molecular basis of compartmentalized intestinal responses to pathogenic insults and may pave the way for novel preventive and therapeutic strategies to mitigate intestinal damage and combat intestinal infections.
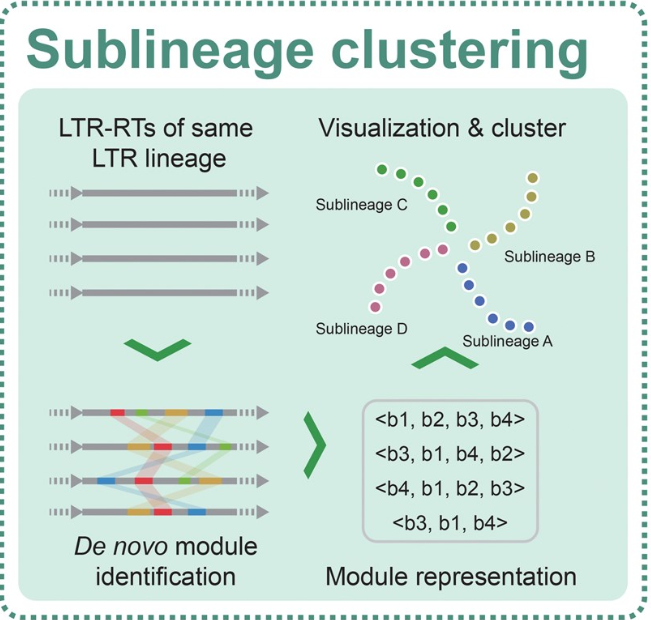
Deciphering Complex Interactions Between LTR Retrotransposons and Three Papaver Species Using LTR_Stream
Tun Xu (徐暾), Stephen J. Bush, Yizhuo Che (车一卓), Huanhuan Zhao (赵焕焕), Tingjie Wang (王庭杰), Peng Jia (贾鹏), Songbo Wang (王松渤), Peisen Sun (孙培森), Pengyu Zhang (张鹏宇), Shenghan Gao (高胜寒), Yu Xu (徐煜), Chengyao Wang (王澄瑶), Ningxin Dang (党宁馨), Yong E. Zhang (张勇), Xiaofei Yang (杨晓飞), Kai Ye (叶凯)
Genomics, Proteomics & Bioinformatics 2025
Long terminal repeat retrotransposons (LTR-RTs), a major type of class I transposable elements, are the most abundant repeat element in plants. The study of the interactions between LTR-RTs and the host genome relies on high-resolution characterization of LTR-RTs. However, for non-model species, this remains a challenge. To address this, we developed LTR_Stream for sublineage clustering of LTR-RTs in specific or closely related species, providing higher precision than current database-based lineage-level clustering. Using LTR_Stream, we analyzed Retand LTR-RTs in three Papaver species. Our findings show that high-resolution clustering reveals complex interactions between LTR-RTs and the host genome. For instance, we found that autonomous Retand elements could spread among the ancestors of different subgenomes, like retroviral pandemics, enriching genetic diversity. Additionally, we identified that specific truncated fragments containing transcription factor motifs such as TCP and bZIP may contribute to the generation of novel topologically associating domain-like (TAD-like) boundaries. Notably, our pre-allopolyploidization and post-allopolyploidization comparisons show that these effects diminished after allopolyploidization, suggesting that allopolyploidization may be one of the mechanisms by which Papaver species cope with LTR-RTs. We demonstrated the potential application of LTR_Stream and provided a reference case for studying the interactions between LTR-RTs and the host genome in non-model plant species.
Deciphering Complex Interactions Between LTR Retrotransposons and Three Papaver Species Using LTR_Stream
Tun Xu (徐暾), Stephen J. Bush, Yizhuo Che (车一卓), Huanhuan Zhao (赵焕焕), Tingjie Wang (王庭杰), Peng Jia (贾鹏), Songbo Wang (王松渤), Peisen Sun (孙培森), Pengyu Zhang (张鹏宇), Shenghan Gao (高胜寒), Yu Xu (徐煜), Chengyao Wang (王澄瑶), Ningxin Dang (党宁馨), Yong E. Zhang (张勇), Xiaofei Yang (杨晓飞), Kai Ye (叶凯)
Genomics, Proteomics & Bioinformatics 2025
Long terminal repeat retrotransposons (LTR-RTs), a major type of class I transposable elements, are the most abundant repeat element in plants. The study of the interactions between LTR-RTs and the host genome relies on high-resolution characterization of LTR-RTs. However, for non-model species, this remains a challenge. To address this, we developed LTR_Stream for sublineage clustering of LTR-RTs in specific or closely related species, providing higher precision than current database-based lineage-level clustering. Using LTR_Stream, we analyzed Retand LTR-RTs in three Papaver species. Our findings show that high-resolution clustering reveals complex interactions between LTR-RTs and the host genome. For instance, we found that autonomous Retand elements could spread among the ancestors of different subgenomes, like retroviral pandemics, enriching genetic diversity. Additionally, we identified that specific truncated fragments containing transcription factor motifs such as TCP and bZIP may contribute to the generation of novel topologically associating domain-like (TAD-like) boundaries. Notably, our pre-allopolyploidization and post-allopolyploidization comparisons show that these effects diminished after allopolyploidization, suggesting that allopolyploidization may be one of the mechanisms by which Papaver species cope with LTR-RTs. We demonstrated the potential application of LTR_Stream and provided a reference case for studying the interactions between LTR-RTs and the host genome in non-model plant species.
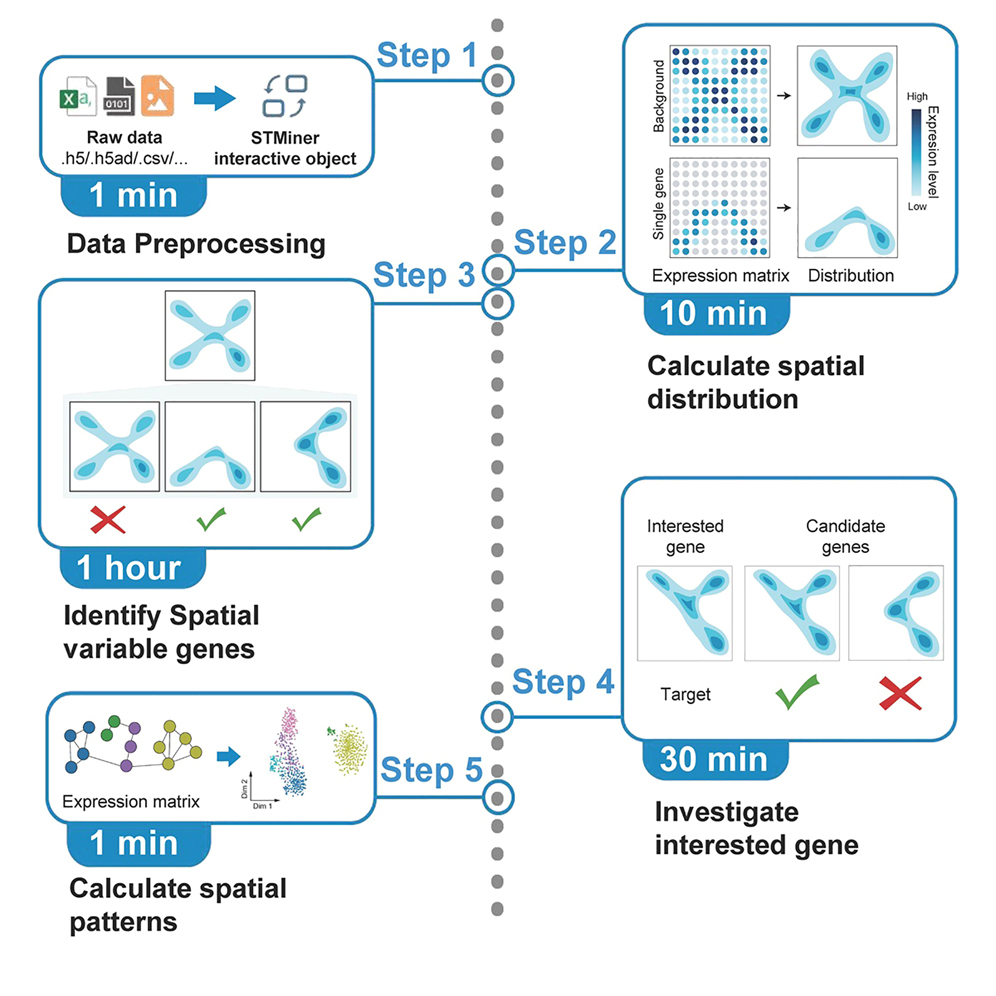
Protocol to decipher complex spatial transcriptomics data using STMiner
Peisen Sun (孙培森)*, Mingxuan Li (黎明轩)*, Kai Ye (叶凯) (* equal contribution)
STAR protocols 2025
Complex spatial transcriptomics (ST) data analysis can be challenging due to uneven sampling, sparsity, and ambiguous tissue boundaries. Here, we present a protocol for deciphering complex ST data using STMiner. We describe steps for installing STMiner, loading ST data into STMiner, and identifying spatially variable genes. We then detail procedures for determining gene sets associated with the structure of interest and obtaining spatial expression patterns. This protocol can be applied to varying resolutions and platforms, without additional reference data.
Protocol to decipher complex spatial transcriptomics data using STMiner
Peisen Sun (孙培森)*, Mingxuan Li (黎明轩)*, Kai Ye (叶凯) (* equal contribution)
STAR protocols 2025
Complex spatial transcriptomics (ST) data analysis can be challenging due to uneven sampling, sparsity, and ambiguous tissue boundaries. Here, we present a protocol for deciphering complex ST data using STMiner. We describe steps for installing STMiner, loading ST data into STMiner, and identifying spatially variable genes. We then detail procedures for determining gene sets associated with the structure of interest and obtaining spatial expression patterns. This protocol can be applied to varying resolutions and platforms, without additional reference data.
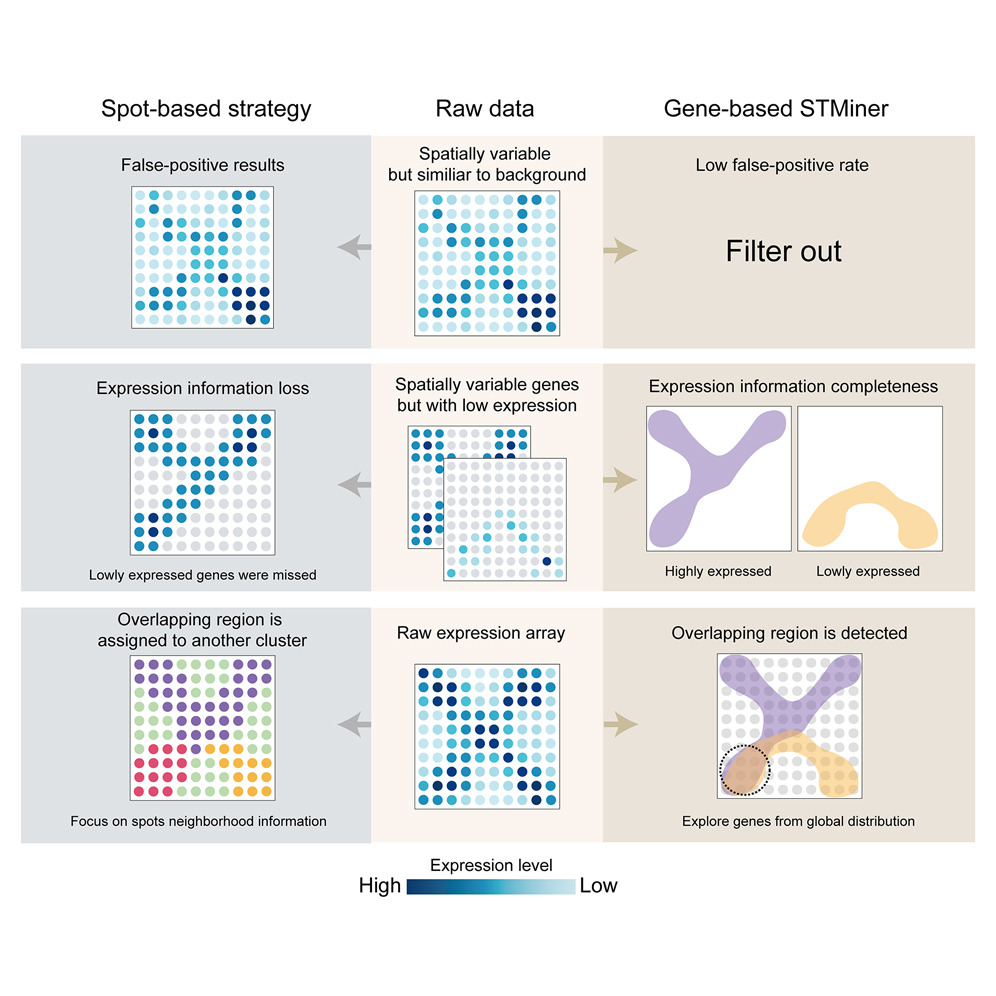
STMiner: Gene-centric spatial transcriptomics for deciphering tumor tissues
Peisen Sun (孙培森), Stephen J. Bush, Songbo Wang (王松渤), Peng Jia (贾鹏), Mingxuan Li (黎明轩), Tun Xu (徐暾), Pengyu Zhang (张鹏宇), Xiaofei Yang (杨晓飞), Chengyao Wang (王澄瑶), Linfeng Xu (许林峰), Tingjie Wang (王庭杰), Kai Ye (叶凯)
Cell Genomics 2025 Featured content
Analyzing spatial transcriptomics data from tumor tissues poses several challenges beyond those of healthy samples, including unclear boundaries between different regions, uneven cell densities, and relatively higher cellular heterogeneity. Collectively, these bias the background against which spatially variable genes are identified, which can result in misidentification of spatial structures and hinder potential insight into complex pathologies. To overcome this problem, STMiner leverages 2D Gaussian mixture models and optimal transport theory to directly characterize the spatial distribution of genes rather than the capture locations of the cells expressing them (spots). By effectively mitigating the impacts of both background bias and data sparsity, STMiner reveals key gene sets and spatial structures overlooked by spot-based analytic tools, facilitating novel biological discoveries. The core concept of directly analyzing overall gene expression patterns also allows for a broader application beyond spatial transcriptomics, positioning STMiner for continuous expansion as spatial omics technologies evolve.
STMiner: Gene-centric spatial transcriptomics for deciphering tumor tissues
Peisen Sun (孙培森), Stephen J. Bush, Songbo Wang (王松渤), Peng Jia (贾鹏), Mingxuan Li (黎明轩), Tun Xu (徐暾), Pengyu Zhang (张鹏宇), Xiaofei Yang (杨晓飞), Chengyao Wang (王澄瑶), Linfeng Xu (许林峰), Tingjie Wang (王庭杰), Kai Ye (叶凯)
Cell Genomics 2025 Featured content
Analyzing spatial transcriptomics data from tumor tissues poses several challenges beyond those of healthy samples, including unclear boundaries between different regions, uneven cell densities, and relatively higher cellular heterogeneity. Collectively, these bias the background against which spatially variable genes are identified, which can result in misidentification of spatial structures and hinder potential insight into complex pathologies. To overcome this problem, STMiner leverages 2D Gaussian mixture models and optimal transport theory to directly characterize the spatial distribution of genes rather than the capture locations of the cells expressing them (spots). By effectively mitigating the impacts of both background bias and data sparsity, STMiner reveals key gene sets and spatial structures overlooked by spot-based analytic tools, facilitating novel biological discoveries. The core concept of directly analyzing overall gene expression patterns also allows for a broader application beyond spatial transcriptomics, positioning STMiner for continuous expansion as spatial omics technologies evolve.
2021
Rcirc: an R package for circRNA analyses and visualization
Peisen Sun, Haoming Wang, Guanglin Li
Frontiers in Genetics 2021
Circular RNA (circRNA), which has a closed-loop structure, is a special type of endogenous RNA that plays important roles in many biological processes. With improvements in next-generation sequencing technology and bioinformatics methods, some tools have been published for circRNA detection based on RNA-seq. Here, we developed the R package “Rcirc” for further analyses of circRNA after its detection. Rcirc identifies the coding ability of circRNA and visualizes various aspects of this feature. It also provides general visualization for both single circRNAs and meta-features of thousands of circRNAs. Rcirc was designed as a user-friendly tool that provides many highly automated functions without requiring the user to perform many complicated processes. It is available on GitHub (https://github.com/PSSUN/Rcirc) under the license GPL 3.0.
Rcirc: an R package for circRNA analyses and visualization
Peisen Sun, Haoming Wang, Guanglin Li
Frontiers in Genetics 2021
Circular RNA (circRNA), which has a closed-loop structure, is a special type of endogenous RNA that plays important roles in many biological processes. With improvements in next-generation sequencing technology and bioinformatics methods, some tools have been published for circRNA detection based on RNA-seq. Here, we developed the R package “Rcirc” for further analyses of circRNA after its detection. Rcirc identifies the coding ability of circRNA and visualizes various aspects of this feature. It also provides general visualization for both single circRNAs and meta-features of thousands of circRNAs. Rcirc was designed as a user-friendly tool that provides many highly automated functions without requiring the user to perform many complicated processes. It is available on GitHub (https://github.com/PSSUN/Rcirc) under the license GPL 3.0.
2019
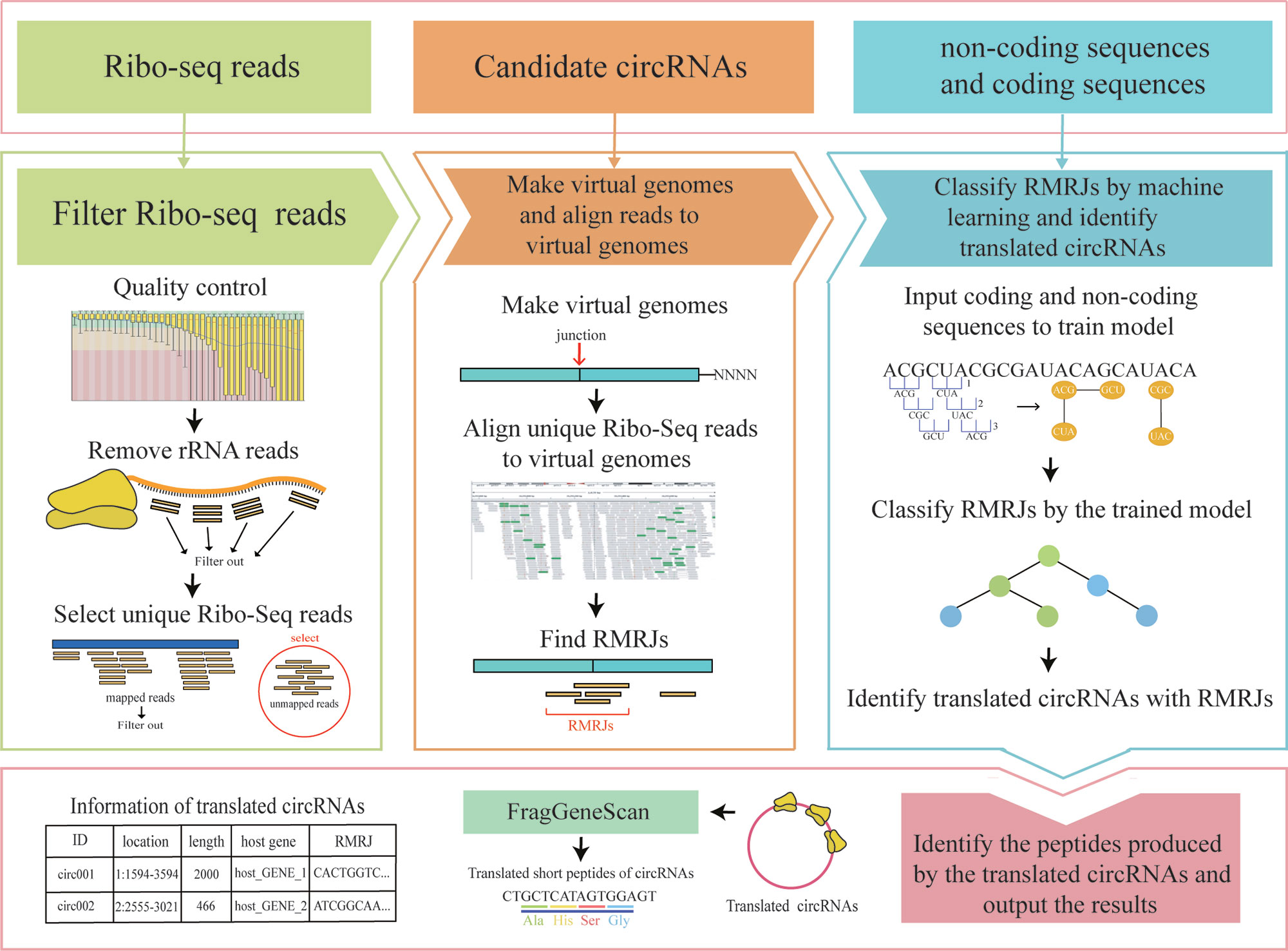
CircCode: A Powerful Tool for Identifying circRNA Coding Ability
Peisen Sun, Guanglin Li
Frontiers in Genetics 2019
Circular RNAs (circRNAs), which play vital roles in many regulatory pathways, are widespread in many species. Although many circRNAs have been discovered in plants and animals, the functions of these RNAs have not been fully investigated. In addition to the function of circRNAs as microRNA (miRNA) decoys, the translation potential of circRNAs is important for the study of their functions; yet, few tools are available to identify their translation potential. With the development of high-throughput sequencing technology and the emergence of ribosome profiling technology, it is possible to identify the coding ability of circRNAs with high sensitivity. To evaluate the coding ability of circRNAs, we first developed the CircCode tool and then used CircCode to investigate the translation potential of circRNAs from humans and Arabidopsis thaliana. Based on the ribosome profile databases downloaded from NCBI, we found 3,610 and 1,569 translated circRNAs in humans and A. thaliana, respectively. Finally, we tested the performance of CircCode and found a low false discovery rate and high sensitivity for identifying circRNA coding ability. CircCode, a Python 3–based framework for identifying the coding ability of circRNAs, is also a simple and powerful command line-based tool. To investigate the translation potential of circRNAs, the user can simply fill in the given configuration file and run the Python 3 scripts.
CircCode: A Powerful Tool for Identifying circRNA Coding Ability
Peisen Sun, Guanglin Li
Frontiers in Genetics 2019
Circular RNAs (circRNAs), which play vital roles in many regulatory pathways, are widespread in many species. Although many circRNAs have been discovered in plants and animals, the functions of these RNAs have not been fully investigated. In addition to the function of circRNAs as microRNA (miRNA) decoys, the translation potential of circRNAs is important for the study of their functions; yet, few tools are available to identify their translation potential. With the development of high-throughput sequencing technology and the emergence of ribosome profiling technology, it is possible to identify the coding ability of circRNAs with high sensitivity. To evaluate the coding ability of circRNAs, we first developed the CircCode tool and then used CircCode to investigate the translation potential of circRNAs from humans and Arabidopsis thaliana. Based on the ribosome profile databases downloaded from NCBI, we found 3,610 and 1,569 translated circRNAs in humans and A. thaliana, respectively. Finally, we tested the performance of CircCode and found a low false discovery rate and high sensitivity for identifying circRNA coding ability. CircCode, a Python 3–based framework for identifying the coding ability of circRNAs, is also a simple and powerful command line-based tool. To investigate the translation potential of circRNAs, the user can simply fill in the given configuration file and run the Python 3 scripts.