
PUBLICATIONS / YEAR
About Me
I am Peisen Sun, a Ph.D. candidate in the Department of Control Science and Engineering at Xi'an Jiaotong University. My research primarily focuses on bioinformatics.
I employ machine learning and artificial intelligence approaches to address complex genomics problems, leveraging state-of-the-art single-cell and spatial omics technologies. My work aims to elucidate the molecular mechanisms underlying biological systems.
My research contributions have been published as first author (including co-first author) in journals such as Cell Genomics and Nature Communications.
Warning
Problem: The current name of your GitHub Pages repository ("Solution: Please consider renaming the repository to "
http://".
However, if the current repository name is intended, you can ignore this message by removing "{% include widgets/debug_repo_name.html %}" in index.html.
Action required
Problem: The current root path of this site is "baseurl ("_config.yml.
Solution: Please set the
baseurl in _config.yml to "Education
-
 Xi'an Jiaotong UniversityPh.D. in Control Science and Engineering
Xi'an Jiaotong UniversityPh.D. in Control Science and Engineering -
 Shaanxi Normal UniversityM.S. in Bioinformatics
Shaanxi Normal UniversityM.S. in Bioinformatics -
 Northwest A&F UniversityB.S. in Bioinformatics
Northwest A&F UniversityB.S. in Bioinformatics
Honors & Awards
-
Outstanding Graduate2021
-
National Scholarship2021
-
Graduate First-Class Scholarship2021
-
Graduate First-Class Scholarship2020
News
Selected Publications (view all )
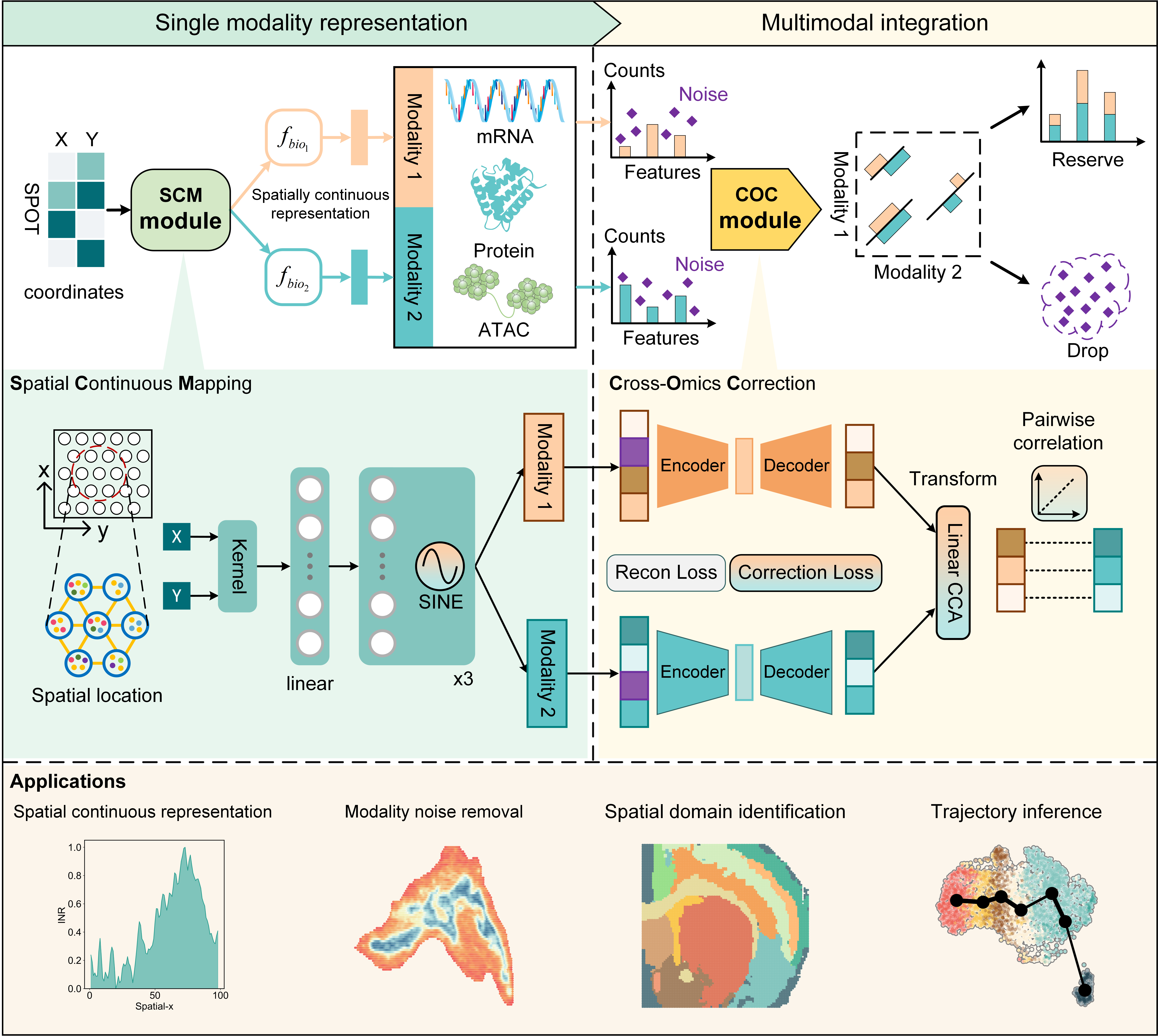
SpatialCOC: an integrative framework for spatial continuous mapping and cross-omics correction in spatial multi-omics data
Mingxuan Li (黎明轩)*, Peisen Sun (孙培森)*, Yisi Luo (罗倚斯), Guancheng Zhou (周冠程), Xiaofei Yang (杨晓飞), Deyu Meng (孟德宇), Kai Ye (叶凯) (* equal contribution)
Nature Communications 2026
Integrating spatial multi-omics data presents significant challenges, particularly in uncovering the spatial patterns of cells and deciphering the real regulatory mechanisms among various omics. These insights are critical for harnessing the full potential of each modality while minimizing the impact of biotechnological biases that will lead to unstable results. Here, we introduce SpatialCOC, a framework that treats spatial information as prior knowledge to learn omics-specific spatial distributions, then discovering nonlinear correlations among modalities. The effectiveness and robustness of SpatialCOC are validated using real-world datasets, encompassing diverse tissue sections analyzed with multiple experimental techniques. Compared to existing methods, SpatialCOC excels in identifying region-specific continuous spatial domains and maintains batch-consistency across trajectory inferences. By providing a novel perspective on the interplay between spatial information and multi-omics modalities, SpatialCOC offers a flexible approach that can accommodate modality data of arbitrary dimensions.
SpatialCOC: an integrative framework for spatial continuous mapping and cross-omics correction in spatial multi-omics data
Mingxuan Li (黎明轩)*, Peisen Sun (孙培森)*, Yisi Luo (罗倚斯), Guancheng Zhou (周冠程), Xiaofei Yang (杨晓飞), Deyu Meng (孟德宇), Kai Ye (叶凯) (* equal contribution)
Nature Communications 2026
Integrating spatial multi-omics data presents significant challenges, particularly in uncovering the spatial patterns of cells and deciphering the real regulatory mechanisms among various omics. These insights are critical for harnessing the full potential of each modality while minimizing the impact of biotechnological biases that will lead to unstable results. Here, we introduce SpatialCOC, a framework that treats spatial information as prior knowledge to learn omics-specific spatial distributions, then discovering nonlinear correlations among modalities. The effectiveness and robustness of SpatialCOC are validated using real-world datasets, encompassing diverse tissue sections analyzed with multiple experimental techniques. Compared to existing methods, SpatialCOC excels in identifying region-specific continuous spatial domains and maintains batch-consistency across trajectory inferences. By providing a novel perspective on the interplay between spatial information and multi-omics modalities, SpatialCOC offers a flexible approach that can accommodate modality data of arbitrary dimensions.
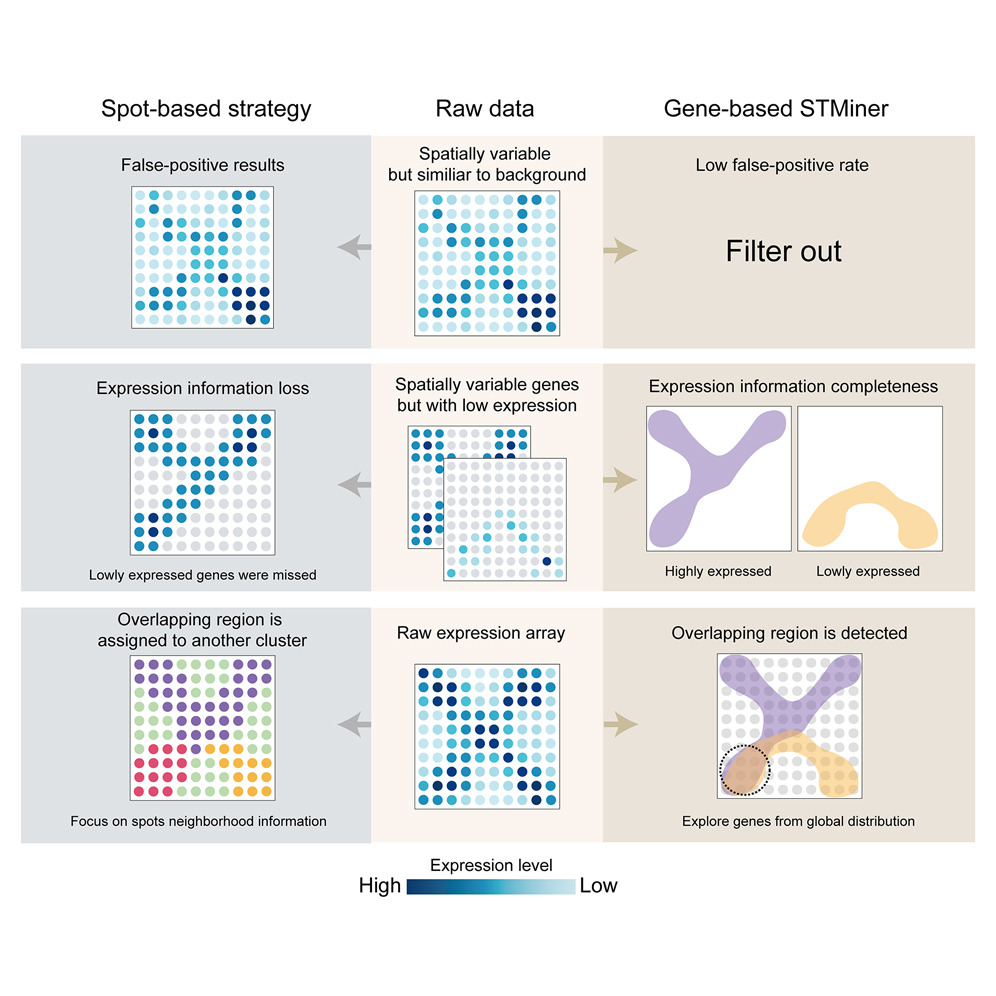
STMiner: Gene-centric spatial transcriptomics for deciphering tumor tissues
Peisen Sun (孙培森), Stephen J. Bush, Songbo Wang (王松渤), Peng Jia (贾鹏), Mingxuan Li (黎明轩), Tun Xu (徐暾), Pengyu Zhang (张鹏宇), Xiaofei Yang (杨晓飞), Chengyao Wang (王澄瑶), Linfeng Xu (许林峰), Tingjie Wang (王庭杰), Kai Ye (叶凯)
Cell Genomics 2025 Featured content
Analyzing spatial transcriptomics data from tumor tissues poses several challenges beyond those of healthy samples, including unclear boundaries between different regions, uneven cell densities, and relatively higher cellular heterogeneity. Collectively, these bias the background against which spatially variable genes are identified, which can result in misidentification of spatial structures and hinder potential insight into complex pathologies. To overcome this problem, STMiner leverages 2D Gaussian mixture models and optimal transport theory to directly characterize the spatial distribution of genes rather than the capture locations of the cells expressing them (spots). By effectively mitigating the impacts of both background bias and data sparsity, STMiner reveals key gene sets and spatial structures overlooked by spot-based analytic tools, facilitating novel biological discoveries. The core concept of directly analyzing overall gene expression patterns also allows for a broader application beyond spatial transcriptomics, positioning STMiner for continuous expansion as spatial omics technologies evolve.
STMiner: Gene-centric spatial transcriptomics for deciphering tumor tissues
Peisen Sun (孙培森), Stephen J. Bush, Songbo Wang (王松渤), Peng Jia (贾鹏), Mingxuan Li (黎明轩), Tun Xu (徐暾), Pengyu Zhang (张鹏宇), Xiaofei Yang (杨晓飞), Chengyao Wang (王澄瑶), Linfeng Xu (许林峰), Tingjie Wang (王庭杰), Kai Ye (叶凯)
Cell Genomics 2025 Featured content
Analyzing spatial transcriptomics data from tumor tissues poses several challenges beyond those of healthy samples, including unclear boundaries between different regions, uneven cell densities, and relatively higher cellular heterogeneity. Collectively, these bias the background against which spatially variable genes are identified, which can result in misidentification of spatial structures and hinder potential insight into complex pathologies. To overcome this problem, STMiner leverages 2D Gaussian mixture models and optimal transport theory to directly characterize the spatial distribution of genes rather than the capture locations of the cells expressing them (spots). By effectively mitigating the impacts of both background bias and data sparsity, STMiner reveals key gene sets and spatial structures overlooked by spot-based analytic tools, facilitating novel biological discoveries. The core concept of directly analyzing overall gene expression patterns also allows for a broader application beyond spatial transcriptomics, positioning STMiner for continuous expansion as spatial omics technologies evolve.
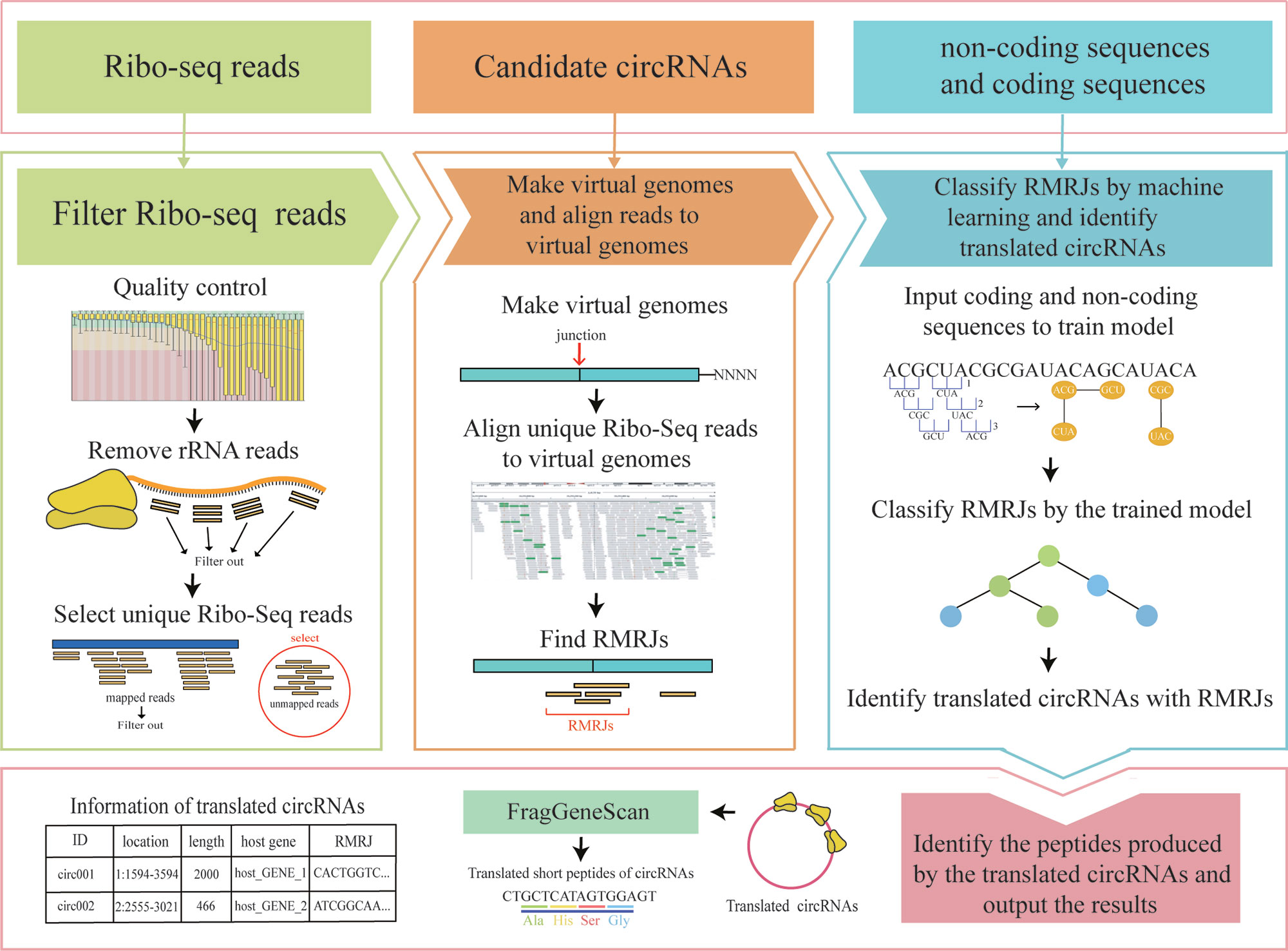
CircCode: A Powerful Tool for Identifying circRNA Coding Ability
Peisen Sun, Guanglin Li
Frontiers in Genetics 2019
Circular RNAs (circRNAs), which play vital roles in many regulatory pathways, are widespread in many species. Although many circRNAs have been discovered in plants and animals, the functions of these RNAs have not been fully investigated. In addition to the function of circRNAs as microRNA (miRNA) decoys, the translation potential of circRNAs is important for the study of their functions; yet, few tools are available to identify their translation potential. With the development of high-throughput sequencing technology and the emergence of ribosome profiling technology, it is possible to identify the coding ability of circRNAs with high sensitivity. To evaluate the coding ability of circRNAs, we first developed the CircCode tool and then used CircCode to investigate the translation potential of circRNAs from humans and Arabidopsis thaliana. Based on the ribosome profile databases downloaded from NCBI, we found 3,610 and 1,569 translated circRNAs in humans and A. thaliana, respectively. Finally, we tested the performance of CircCode and found a low false discovery rate and high sensitivity for identifying circRNA coding ability. CircCode, a Python 3–based framework for identifying the coding ability of circRNAs, is also a simple and powerful command line-based tool. To investigate the translation potential of circRNAs, the user can simply fill in the given configuration file and run the Python 3 scripts.
CircCode: A Powerful Tool for Identifying circRNA Coding Ability
Peisen Sun, Guanglin Li
Frontiers in Genetics 2019
Circular RNAs (circRNAs), which play vital roles in many regulatory pathways, are widespread in many species. Although many circRNAs have been discovered in plants and animals, the functions of these RNAs have not been fully investigated. In addition to the function of circRNAs as microRNA (miRNA) decoys, the translation potential of circRNAs is important for the study of their functions; yet, few tools are available to identify their translation potential. With the development of high-throughput sequencing technology and the emergence of ribosome profiling technology, it is possible to identify the coding ability of circRNAs with high sensitivity. To evaluate the coding ability of circRNAs, we first developed the CircCode tool and then used CircCode to investigate the translation potential of circRNAs from humans and Arabidopsis thaliana. Based on the ribosome profile databases downloaded from NCBI, we found 3,610 and 1,569 translated circRNAs in humans and A. thaliana, respectively. Finally, we tested the performance of CircCode and found a low false discovery rate and high sensitivity for identifying circRNA coding ability. CircCode, a Python 3–based framework for identifying the coding ability of circRNAs, is also a simple and powerful command line-based tool. To investigate the translation potential of circRNAs, the user can simply fill in the given configuration file and run the Python 3 scripts.